Decent Dancing Gentleman
Ok, so here we are taking the first steps in the Gaussian09 program. There is so much to say about it and at the same time so little.
Just for the protocol I do not consider myself to be a super-professional quantum chemist, therefore I cannot take any responsibility for the correctness of that or another approach\basis\method that I’m using. Keep in mind, please.
Here I will present you the input and the output of the calculation in Gaussian. It was done just for fun and the output is incorrect, so please do not use it to derive any conclusions.
Let’s begin with the input. I used the common B3LYP functional with the simple 6-31g basis just to make the calculation time shorter. The molecule is uncharged naturally and its multiplicity equals 2 (because of reasons). Usually for the organic molecules the multiplicity is 1. Now I need to gain the coordinates of the atoms in my complex molecule. As a matter of fact, I despise Gaussian View for its drawing technique and prefer to use Avogadro instead. It is freeeeeeeeeeeeeeee.
Next step is the final step: now I need to specify the command. In
this particular case I didn’t want the geometry to be disturbed for
the sake of design, so I only specified the freq command, which
(unbelievable) makes Gaussian to calculate the frequencies.
Actually, it is a very important step, because, as far as I know, it
helps to understand, whether the geometry of the molecule was
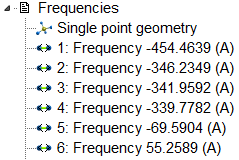
calculated correctly or not. If there are negative frequencies, the
geometry optimisation went wrong (as in this case).
This is the full input script to run in Gaussian 09
#!/bin/csh
g09 <<END >chel.log
%%NProcShared=3
%chk=chel.chk
#n freq b3lyp/6-31g
chelovek
0 2
C 2.59748 1.11074 0.03228
C 3.38462 -0.04548 -0.00429
C 2.78903 -1.31139 -0.04094
C 1.20005 1.00805 0.03148
C 1.39219 -1.43214 -0.03992
C 0.60037 -0.26708 -0.00427
O 0.46502 2.14838 0.06275
O 0.84506 -2.67509 -0.07143
H -0.47671 -0.35096 -0.00529
N -0.46979 -2.90451 -0.06993
N -0.86889 2.16497 0.08375
O 3.55684 -2.41919 -0.07644
O 3.18115 2.32553 0.06912
Ti 5.49705 0.11926 -0.00106
H 4.14329 2.42739 0.07238
H 4.52384 -2.37572 -0.07744
H -1.16274 3.16672 0.06613
H -1.18627 1.76271 0.99562
H -0.60121 -3.94077 -0.06927
H -0.87059 -2.5319 -0.96107
END
echo "Job done. "
Now we should wait for the output. Aaand…after 14 minutes I’ve received the decent gentleman with head made of Titanium and other noticable features, who is more than happy to dance.
There are many other interesting things in output, for example, the enthalpies, entropies etc, but I will write about it some other time. And for now, I should only point out, that the amount of negative frequencies demonstrates the inaccuracy of the geometry optimisation.
THAT’S WHAT MAKES US A GREAT COUNTRY.
THE LITTLE THINGS ARE SERIOUS AND THE BIG ONES ARE NOT.
WILL ROGERS
Job cpu time: 0 days 0 hours 14 minutes 30.0 seconds.